

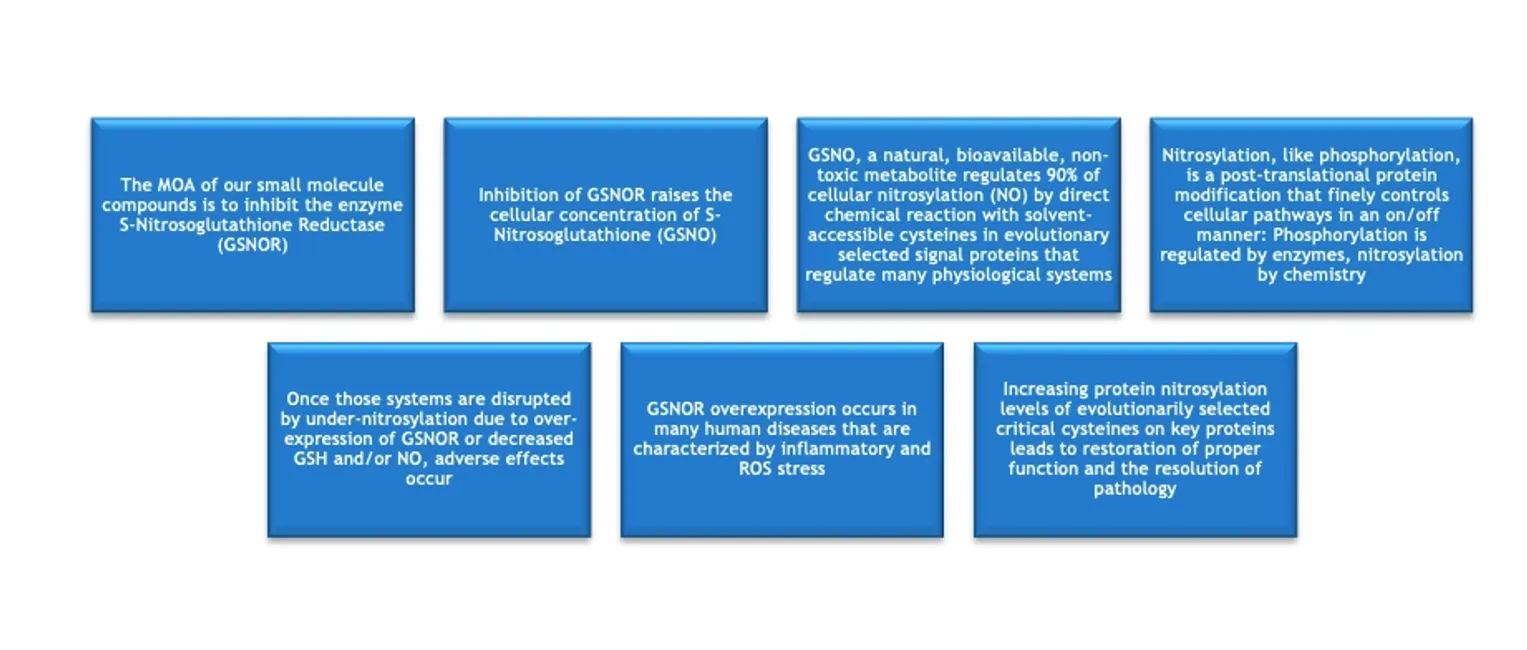

As support for these hypotheses, GSNO Therapeutics and its collaborators have demonstrated that inhibiting the enzyme S-nitrosoglutathione reductase (GSNOR) increases the levels of S-nitrosylation on critical proteins and produces numerous therapeutic benefits that are relevant to many important diseases across multiple organ systems. GSNOR is a reductase that breaks down or reduces S-nitrosoglutathione (GSNO), which is the stable, cellular storage form of S-nitrosylating activity, performing 90% of all cellular nitrosylation. GSNO has a half-life of 5.5 hours in the body and does not break down to NO, but, like NO, nitrosylates cysteines on evolutionarily selected signals and other proteins. NO, in contrast, is extremely reactive with a tissue half-life of seconds and limited tissue penetration, whereas GSNO’s longer half-life and extensive cellular and plasma distribution make it the carrier of the major S-nitrosylating activities. NO reacts as an SN1 type reaction, whereas GSNO uses a different SN2 type of chemical reaction. Both compounds S-nitrosylate solvent accessible cysteines on evolutionarily selected proteins, thereby changing the structure and function of those proteins. Thus, GSNO is the “real API” of precision-targeted GSNORIs.
GSNO itself is a natural metabolite that has been shown to be non-toxic in animal studies and is freely bioavailable in blood and body tissues, and can move from the intestine or other tissues into many organs, including hard-to-access brain ocular spaces. Thus, GSNO is the “real” Active Pharmaceutical Ingredient (API) of GSNORi technology that does the actual therapeutic work by nitrosylating under-nitrosylated proteins in signal pathways to effect therapy. GTI-850 does not directly affect any of the MOAs that it produces. Rather, GSNO does that.
Many human diseases and animal models have increased GSNOR, which can be normalized by inhibiting GSNOR. But, GSNORi is still active even without GSNOR overexpression. Like phosphorylation, nitrosylation is a post-translational protein regulatory system, but one regulated by only one enzyme, GSNOR, rather than by 700+ as with phosphorylation. Therefore, nitrosylation regulation by GSNORi is a much more druggable technology than phosphorylation regulation by so many often overlapping kinase and phosphatase targets, many of which produce toxicity, which GSNORi does not.
Our lead molecule, GTI-850, is a composition of matter compound that we invented and own all rights to. It has no toxicity in sub-chronic safety studies or in vitro studies against 44 common targets of toxicity. GTI-850 has an unusual PK/PD relationship. It has only 0.2% oral bioavailability, yet 20-40% oral bioactivity compared to its IV bioactivity. Why? The answer is that oral GTI-850, while not systemically distributed, inhibits GSNOR in the intestine, which raises GSNO concentrations there, and GSNO moves into the blood and circulates throughout the body and into many organs, including the brain and other neural tissues, where it nitrosylates undernitrosylated proteins for therapeutic benefits.
The intestine expresses high concentrations of GSNOR over its entire surface area, which is huge and communicates directly with the blood circulation. The lack of systemic circulation of GTI-850, along with potent bioactivity, greatly increases the probability of clinical success because none of the typical hazards apply that cause drugs to fail in safety or produce a low therapeutic risk/benefit equation for even marketed drugs. Because GTI-850 remains in the intestine, it will not have the following typical systemic safety issues: no first-pass metabolism, no toxic metabolites, no drug-drug interactions, no inhibition of liver CypP450s, no inadequate PK, no tissue accumulation, and no off-target toxicity. The drug is active with a single dose for acute diseases and at once-a-day dosing in chronic animal disease models.
GSNOR inhibition increases the nitrosylation of evolutionarily selected signal transduction proteins by increasing the cellular concentration of S-nitrosoglutathione (GSNO), which trans-nitrosylates accessible regulatory cysteines. Trans-nitrosylation by GSNO proceeds by a different chemical mechanism than nitrosylation by NO. GSNO does not produce. NO, as some literature suggests. Furthermore, GSNOR inhibition does not cause nitrosative stress but rather prevents it [6,9,10]. This lack of nitrosative stress is a clear advantage for using GSNOR inhibition rather than exogenous NO itself or non-specific NO donors to affect the therapeutic benefits of nitrosylation.
By inhibiting GSNOR, we simultaneously activate many signal pathways in a natural and balanced way to promote multi-faceted therapeutic outcomes, as noted above. This balanced approach is in contrast to drugs that inhibit or inactivate a single target that is disease-related—TNF-α, TGF-β, IL-6, and many others. The evidence shows that our GSNOR inhibition approach is more efficacious and safer because we are simultaneously regulating many disease processes in a balanced way, not obliterating a single disease driver that may also have other important physiological roles. While single effect drugs, such as MABs, have therapeutic efficacy, we believe that our multi-effect drugs will be both safer and more effective for many diseases with multiple and complex pathophysiological drivers—those of primary medical need today.
